
Yesterday I wrote about why negative controls are important in a genome-scale search for functional DNA. Today, I’ll discuss the main focus of our recent work: understanding what makes a piece of DNA functional.
The particular DNA I’m interested in is known by not very functional term ‘cis-regulatory’ DNA – a term that requires six syllables, an italicized Latin prefix, and a hyphen. This is DNA that is crucial in gene decisions: cis-regulatory DNA helps to control when, where, and how much genes are expressed. This happens because cis-regulatory DNA serves as a landing pad for ‘transcription factors’, proteins that land on cis-regulatory DNA and control the expression of nearby (or sometimes not so nearby) genes.
The question that haunts me is this: why don’t transcription factors get lost? My worry follows from these three observations:
1. Transcription factors recognize very short segments of DNA. To give you an example, the transcription factor I study, the eye development factor Cone-rod homeobox (Crx), recognizes slight variations of the 8-base sequence CTAATCCC.
2. Large eukaryotic genomes are packed with millions of copies of these short sequences. The 8-base Crx recognition site occurs more than 6.6 million times in the mouse genome.
3. Only a small fraction of all potential transcription factor recognition sequences are actually bound by transcription factors. In the case of Crx, only about 14,000 of 6.6 million 8-base recognition sequences are bound by Crx in the genome.
I’ve illustrated the problem below. On the left is an image of the transit of Venus across the Sun. On the right, the blue circle shows the small fraction of Crx recognition sites that are bound.

So yes, I’m serious – why don’t transcription factors get lost in giant genomes?
There are two primary answers (not mutually exclusive) that people typically turn to:
Hypothesis A: Chromatin context is everything. In any given cell, most of the genome is inaccessible, wrapped up into large, compact regions of dense chromatin. This reduces the transcription factors’ search space, so they don’t get lost.
But if context is the answer, how do the right parts of the genome get left exposed?
Hypothesis B: DNA grammar provides specificity. Short 8-base pair recognition sequences are not enough; true functional sites consist of rare, highly specific combinations of short recognition sites. The millions of spurious sites in the genome do not have the right DNA grammar, and they are not bound.
But if grammar is the answer, why do transcription factors seem to non-specifically bind all over the place?
We set out to resolve this dilemma by testing the cis-regulatory function of two types of genomic DNA:
Bound DNA: DNA segments with Crx recognition sites that are bound by Crx in the genome (i.e., ChIP-seq peaks).
Unbound DNA: DNA segments with equal numbers of good Crx recognition sites, but which are not bound by Crx in the genome.
Importantly, we tested these DNA pieces outside of their native genomic context, in the very permissive context of plasmids.
So, under Hypothesis A, both bound DNA and unbound DNA should be functional, because they have both been removed from any genomic context and placed on plasmids. The context is now the same for both classes of DNA.
Under Hypothesis B, bound DNA should be more functional than unbound DNA, regardless of context.
We tested large numbers of bound and unbound DNAs in our massively parallel1 functional (i.e., reporter gene) assay. (We did this in the dissected whole retinas of baby mice.) The result came as a surprise to me, because I was betting on Hypothesis A. But it turns out that bound DNA differed from our random DNA distribution (showing function), while the unbound DNA largely resembled the random DNA distribution (showing non-function).
You can see this in these histograms from Fig. 1 of our paper. In the first panel, bound DNA distribution is in blue, the random DNA distribution is in green, and the level of gene expression is shown on the x-axis:

In the next panel, unbound DNA is in blue, and random DNA is in green – notice that these two largely overlap (except in the left tail):
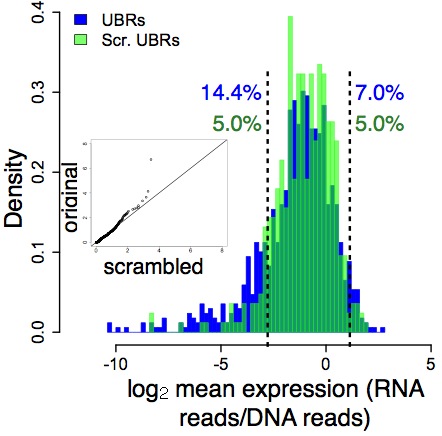
What this means is that the distinction between functional (bound) and non-functional (unbound) DNA is independent of context, at least to a large degree. The information that distinguishes function from non-function is therefore locally encoded in the short DNA regions (84 bases) that we tested.
So the answer must be hypothesis B, DNA grammar, right? Well, maybe, but so far we have been unable to find any obvious grammar. We’re now looking at more subtle structural DNA features that could account for the difference between function and non-function.
One final point – without our random DNA negative controls, our empirical null distribution, we would have drawn a very different conclusion. Unbound DNA would have looked very functional, just not quite as functional as bound DNA. But in fact the unbound DNA, which has lots of Crx recognition motifs, behaves much like our random DNA, which has very few Crx recognition motifs, and that means that most of what the unbound DNA is doing is non-specific, the result perhaps of biological noise.
Our paper: “Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks”, PNAS July 16, 2013 vol. 110 no. 29 11952-11957.
1. Yes, ‘massively parallel’ is a technical term, one that has a different meaning from ‘high thoughput’.

Could non-specific binding of transcription factors be a mechanism for regulating the pool of available transcription factors?
That’s one idea that’s out there – the ‘molecular clutch’ hypothesis, or the TF depot hypothesis, right? Non-specific binding must be hard to avoid.
On the other hand, I expected to see in my experiment a lot of ChIP-seq peak sequences that were the result of non-specific but reproducible binding. One third of the ChIP-peaks I tested had no Crx motif in them, suggesting that these are the result of non-specific binding. And yet those sequences were also strong activators in the assay.
There are still many outstanding questions.
Do you have RSS feed?
We did have an icon at one point… here’s the URL: http://feeds.feedburner.com/thefinchandpea/HSUg
As Mike, noted, we do. Hopefully, the buttons will be back up in a few minutes. Our ne’er-do-well, managing editor bought a house and Time Warner has continually delayed his acquisition of internet. Fortunately, Mike requires no editing.
Nice work. Do you think that the local grammar is having a larger impact on actual Crx binding, or the functional outcome driven by successfully recruiting other proteins? In your comment above, you say that some of the ChIP peaks don’t have a Crx motif at all. This seems to scream indirect association through other interaction with other DNA-binding proteins more than nonspecific binding of Crx itself…
Thanks! Indirect association is one possibility, but my hunch is that many of the ChIP peaks without Crx motifs are reproducible-but-non-specific binding to autonomously functional sequences. This is because many of the ChIP peaks lacking Crx motifs have all of the following traits:
– they fall in CpG islands
– they sit within 1 kb of a TSS
– they fall in non-cell-type-specific DNase I hypersensitive sites
– they fall in TF-binding hotspots (i.e. many ChIP peaks for other factors in other cell type fall there)
I now have data from Crx-/- retina, not yet analyzed, but it will tell us whether these ChIP peaks without motifs lose activity when you take away the TF. If they do, then it’s probably indirect association.
Very enlightening paper, indeed. Thanks !
Two questions: 1. What exactly do you mean by DNA grammar ? Sequence motifs flanking the binding site ? GC % ?
2.What, in your opinion, is the extent to which these results can be generalized to other sequence-specific TFs ?
Sorry for the delay – somehow I missed your comment. With DNA grammar, I mean ways of arranging DNA sequence into combinations that produce functional cis-regulatory elements – usually TF binding motifs, but I’m also investigating the role of other structural features that have some sequence degeneracy.
I do think the CRE-seq assay results are likely to generalize well – I’ve no reason to think Crx is special as far as concerns its relationship between binding and genomic context.
To validate the grammar hypothesis then, do you have a dataset where the motifs in the CBRs are intact but the rest of the sequence is scrambled ?
That’s a good experiment to do, but right now we think that the grammar involves more than just the Crx motifs, so we expect that scrambling the intervening region would kill much of the activity.
We’re currently designing a new library aimed at testing several different grammar hypotheses. Our experiment will include what you suggested, and also cases we try to turn a poorly expressing sequence into a highly expressing one by altering the DNA structural features (minor groove width, propeller twist, etc.) flanking the motifs.