
Last September, there was a wee bit of a media frenzy over the Phase 2 ENCODE publications. The big story was supposed to be that ‘junk DNA is debunked’ – ENCODE had allegedly shown that instead of being filled with genetic garbage, our genomes are stuffed to the rafters with functional DNA. In the backlash against this storyline, many of us pointed out that the problem with this claim is that it conflates biochemical and organismal definitions of function: ENCODE measured biochemical activities across the human genome, but those biochemical activities are not by themselves strong proof that any particular piece of DNA actually does something useful for us.
The claim that ENCODE results disprove junk DNA is wrong because, as I argued back in the fall, something crucial is missing: a null hypothesis. Without a null hypothesis, how do you know whether to be surprised that ENCODE found biochemical activities over most of the genome? What do you really expect non-functional DNA to look like?
In our paper in this week’s PNAS, we take a stab at answering this question with one of the largest sets of randomly generated DNA sequences ever included in an experimental test of function. We tested 1,300 randomly generated DNAs (more than 100 kb total) for regulatory activity. It turns out that most of those random DNA sequences are active. Conclusion: distinguishing function from non-function is very difficult.
To test DNA for function, we used a new technique to measure whether a piece of DNA can regulate a downstream gene (a barcoded DsRed reporter gene). One way to define functional DNA in the context of this experiment is ‘any piece of DNA that reproducibly regulates the reporter gene.’
We tested about 2,000 native sequences from the genome (more about that in my next post), and, as a negative control, we also tested random DNAs, DNAs created by scrambling the sequences of genomic DNA.
It turns out that most of the 1,300 random DNA sequences cause reproducible regulatory effects on the reporter gene. You can see this in these results from 620 random DNA sequences below, in what I call a Tie Fighter plot:

A histogram of gene expression level is shown by the green bars (gene expression is the x-axis). You can see that the 620 random DNA sequences cover a wide range of gene expression. But, crucially, this wide range of expression is not technical variance. The black Tie Fighter things show the results for five individual random DNAs (dot shows the mean, bars are +/- 95% C.I.). They show that different random DNAs have distinct and reproducible regulatory effects.
In other words, it’s very easy to generate a random piece of DNA that looks functional.
These results show why we need a more stringent definition of function. In our experiment, the distribution of random DNA activities is an empirical null distribution – a genome-scale control for a genome-scale functional assay. This distribution shows us how random DNA behaves in the experiment. If a class of DNA elements deviates from that random distribution, then it’s surprising and we call it functional. We compared the activities of putatively functional DNA sequences (ChIP-seq peak regions, blue) against the random DNA (green):
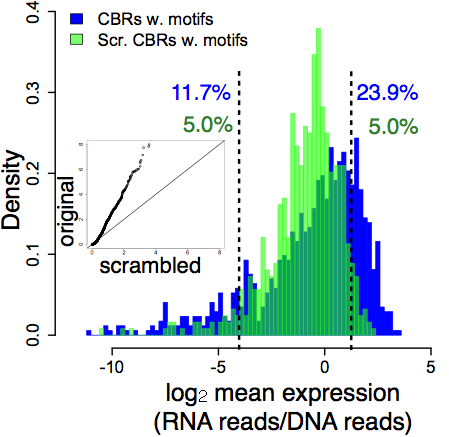
The big lesson here is perhaps not surprising – most DNA will look functional at the biochemical level. The inside of a cell nucleus is a chemically active place. The real puzzle is this: how does functional DNA manage to distinguish itself from the vast excess of dead transposable elements, pseudogenes, and other accumulated junk?

Thanks for the summary. I like the approach that you took… it’s especially illuminating in light of the ENCODE hype.
My biggest problem with the ENCODE publications in the fall was the hype over the ‘junk DNA is debunked’ idea, which is just flat-out wrong. It’s unfortunate because this story obscured other, more valuable aspects of ENCODE.
Great study. If I read the paper correctly, the only obvious sequence difference between the unbound and bound sequences is GC content? Have you tried increasing the GC content around the binding motifs in the unbound sequences? I wonder if that would change their level of function. What other features could be conferred by the sequence alone…secondary structure?
Great question! We are currently trying to convert non-functional to functional sites by changing the surrounding GC content. There are particular DNA structural features that we’re looking at, like minor groove width, propeller twist, etc. that could potentially explain the difference between bound and unbound motifs.
“The real puzzle is this: how does functional DNA manage to distinguish itself from the vast excess of dead transposable elements, pseudogenes, and other accumulated junk?”
What do you mean by “distinguish itself”? How do we know it is distinguishing itself as being different in any way other than in how we think of it in our minds? (Sorry if this is a naive question; I am genuinely curious as to what you mean.)
I just mean that a typical functional factor binding site is very small, and the genome is very large. Those small binding sites need to attract regulatory proteins that could potentially stick to other (non-functional) places in the genome.
Ahh, got it. That makes sense. Thanks!
The approach you took in this study is very nice, and I think that you are asking the right question. Good job!! I am a little puzzled, still, by the last sentence of the paper “Our results show that the cis-regulatory potential of TF-bound DNA is determined largely by highly local sequence features and not by genomic context”. You obviously don’t expect to see an effect of the genomic context on a plasmid, but does that mean there is none in the genome? Actually, there is ample evidence of the opposite in PEV and transgene silencing. But perhaps I don’t understand it right.
You make a great point. Our last sentence was intentionally provocative. No question, genomic context would make a difference if we integrated these constructs into different places in the genome. (In fact, we’re gearing up to do this.) But I argue that the difference between and non-function is not due to context.
What I mean is this: I predict that the ChIP peak sequences (as a class) will show more activity than random DNA, while the unbound sequences (as a class) will not, regardless of genomic context (except in cases where everything is completely repressed by context).